


A Food and Drug Administration (FDA) investigation alleges that for years, the Philips Respironics company knew of health risks associated with its sleep aid devices but did not immediately act. NBC 5 Responds’ Lisa Parker reports.
Millions of people across the country were jostled awake this year over fearful news: A critical device they were prescribed and relied on each night for sleep was suddenly labeled a hazard to their health.
A massive voluntary recall suddenly took many sleep aid devices off the market without offering a replacement.
Now, newly released documents are raising questions over whether the company at the center of that recall knew of the problems and risks with its devices for years, but did not immediately act.
For those diagnosed with sleep apnea, a condition where a person can stop breathing while sleeping, the Philips Respironics’ CPAP, BiPAP and ventilator machines were the most popular and often the go-to choice for doctors prescribing a sleep aid device.
Feeling out of the loop? We'll catch you up on the Chicago news you need to know. Sign up for the weekly Chicago Catch-Up newsletter here.
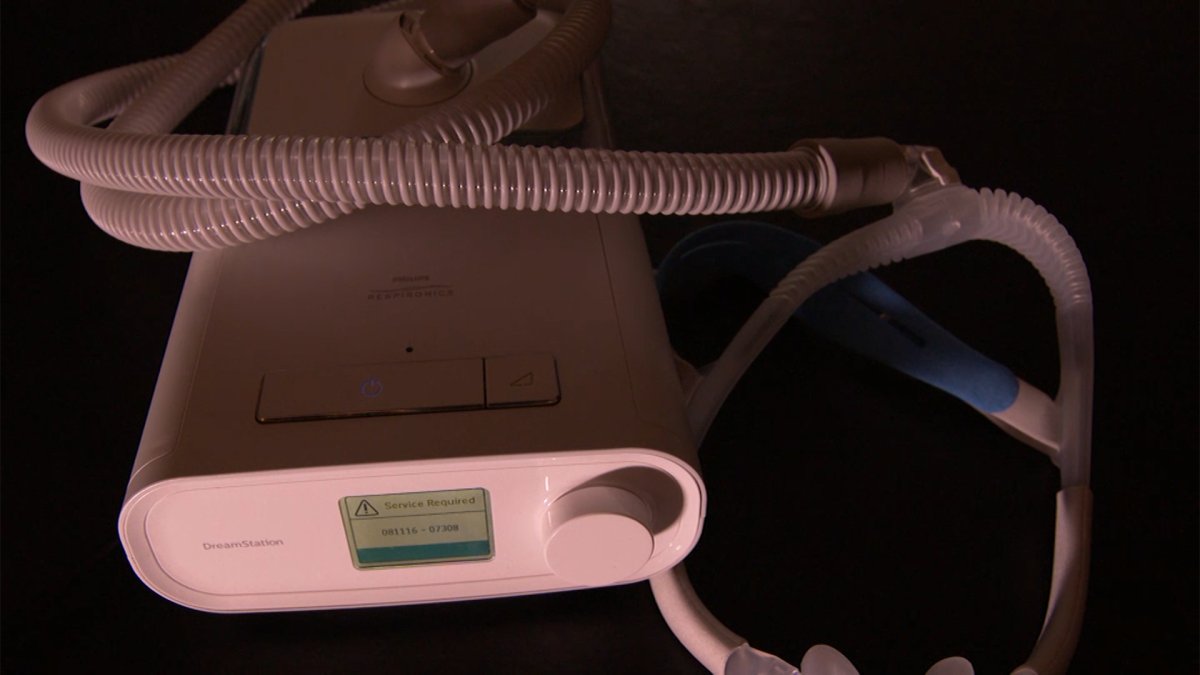
That’s why it came as a shock when Philips Respironics announced a voluntary recall of more than 15 million devices in June. Half of those devices are in use in the U.S., the company said previously.
The reason behind the recall: a noise abatement foam used inside the machine could degrade, and if inhaled, the user faced a serious risk of injury, or worse, cancer.
So many of these Philips devices were recalled that there weren’t enough resources or machines to offer patients a repair or replacement for up to a year, the company previously told NBC 5 Responds.
I depend on it to sleep at night… This is life altering.
Terri Domingo, Philips CPAP Device User
While the recall was a startling surprise for users of these devices, the problems at the heart of the matter were not a surprise to the company, as it may have known of the issues for quite some time, according to an investigation by the Food and Drug Administration (FDA).
The voluntary Class 1 recall of the devices triggered several FDA inspections of Philips Respironics’ manufacturing facility in Pennsylvania this past August and November.
Among many things, the FDA inspectors looked through the company’s digital records, facility operations and product testing, all in an effort to determine what may have caused or contributed to the foam issues that led to the recall and assess whether the company had met federal manufacturing requirements.
In example after example, investigators allege for years, staff within the Philips Respironics company knew of foam degradation problems in its devices but did not immediately act. In addition, the investigation found executive managers for the company knew of the problems for more than a year before issuing the voluntary recall.
According to the FDA’s inspection, there were warnings from inside the company. This included "at least 14 tests or assessments" from as early as 2016 where Philips’ staff were “aware of issues and concerns related to potential foam degradation.”
There were internal emails that FDA inspectors cite including one in 2015 from an employee of Philips to the company’s raw foam supplier that “implies that a customer made [Phillips] aware of polyester polyurethane foam degradation issues,” the report said.


After the company had received complaints of foam degradation in its products, in 2018, inspectors found a conversation over email with Philips’ staff acknowledging the company’s own internal tests had confirmed the foam could fall apart when exposed to high humidity or heat.
But, the FDA inspection found, in that same conversation, Philips’ personnel said the company had “made the decision to not change the design” and continue to include the foam in its ventilator devices.
And from outside of the company’s walls, inspectors found there were external warnings, including tens of thousands of consumer complaints that the FDA said did not trigger any immediate action.
An analysis of consumer complaints sent to the company revealed that since 2008, Philips Respironics had received "over 222,000 complaints" that included the keywords "contaminants, particles, foam, debris, airway, particulate, airpath and black."
Of those, at least 110 complaints from 2014 to 2017 were directly related to the foam degradation problems at the heart of the voluntary recall.
The FDA’s inspection found Philips’ Executive Management were made aware of potential foam degradation from "as early as January of 2020, or earlier, but implemented no corrective actions until April 2021."
FDA inspectors said, "Additionally, [Philips Respironics] became aware of this [foam degradation] issue and related field complaints in at least 2015 or earlier."
When asked for comment, Philips Respironics referred NBC 5 Responds to a prepared list of FAQs that responded to some, but not all, of the FDA’s findings, as well as a statement from the Royal Philips CEO Frans van Houten (Philips Respironics is a subsidiary of Royal Philips).
"We will work closely with the FDA to clarify and follow up on the inspectional findings…" Van Houten said in part. "Until we have concluded these discussions, we are not able to publicly provide further details."
In response to the consumer complaints the FDA uncovered, Philips said, "In prior years, there were limited complaints related to foam degradation, which were evaluated and addressed on a case-by-case basis."
The company added that problems involving "volatile organic compound (VOC) emissions" or chemicals that could impact a user's health, "started to surface more recently... leading to the actions in the first half of 2021."
Somebody’s got to be held responsible for this.
Shawn Woodruff, Philips CPAP User
The FDA said the findings of its inspection and investigation are not considered a "final determination." Regulators will review Philips’ response to these findings before taking next steps, the FDA said, including determining whether violations occurred.
Questions in the FDA’s investigation also centered around the devices the Philips Respironics company has issued in its ongoing effort to repair or replace all of the machines under recall.
After the initial recall, regulators said Philips Respironics planned to repair the CPAP and BiPAP devices’ polyester-based polyurethane foam – the foam that degrades and can cause injury – with a different, silicone-based foam.
Now, months later, the FDA warns that the new silicone-based foam failed a safety test, and could be hazardous.
"During the manufacturing facility inspection, the FDA obtained additional information, not previously available to the agency, regarding the silicone-based foam used in a singular, similar device marketed outside the U.S., which failed one safety test for the release of certain chemicals of concern," a news release that accompanied the FDA inspection’s findings reads.
At this time, the FDA said it does not have enough information to recommend users stop using those new devices.
"The FDA does not recommend that patients who have participated in the repair and replace program discontinue use of their product," the news release states. "The FDA has reached this determination based on an overall benefit-risk assessment."
Philips Respironics said the failed safety test was for a device that has not "been placed on the market globally" and that previous testing of the replacement device "was previously submitted to the FDA and demonstrated acceptable results."

The FDA's report also calls into question whether there could be more devices that are currently in use containing the hazardous foam that were not included in the company’s voluntary recall.
Inspectors found "no documented investigation, risk analysis or design analysis to support Philips’ rationale for which polyester polyurethane foam-containing products were included or not included in the ongoing recall."
The recent news of what the FDA found inside Philips Respironics is not putting patients at ease.
Some Phillips Respironics device users, including those now part of a class-action lawsuit filed against the company over the recall, believe it needs to be held accountable.
"The lack of communication once they discovered what was wrong with these machines is just criminal," said Shawn Woodruff of McHenry County in Illinois. "That’s terrible to do to people."
Woodruff discovered the recall on his own earlier this year and told NBC 5 Responds he had a difficult time finding a replacement for his Philips CPAP machine.
Woodruff said the situation has left him feeling less in control of what’s going on with his own health.
"You’re messing with [people’s] lives," Woodruff said. "Somebody’s got to be held responsible for this."
Below is a complete list of the Philips Respironics machines that are impacted, according to the FDA:
CPAP and BiPAP Devices
| Device Type | Model Name and Number (All Serial Numbers) |
| Continuous Ventilator, Minimum Ventilatory Support, Facility Use | E30 (Emergency Use Authorization) |
| Continuous Ventilator, Non-life Supporting | DreamStation ASV, DreamStation ST - AVAPS, SystemOne ASV4, C-Series ASV, C-Series S/T and AVAPS, OmniLab Advanced+ |
| Noncontinuous Ventilator | SystemOne (Q-Series), DreamStation, DreamStation Go, Dorma 400, Dorma 500, REMstar SE Auto |
Ventilators
| Device Type | Model Name and Number (All Serial Numbers) |
| Continuous Ventilator | Trilogy 100, Trilogy 200, Garbin Plus, Aeris, LifeVent |
| Continuous Ventilator, Minimum Ventilatory Support, Facility Use | A-Series BiPAP Hybrid A30 (not marketed in the US), A-Series BiPAP V30 Auto |
| Continuous Ventilator, Non-life Supporting | A-Series BiPAP A40, A-Series BiPAP A30 |





